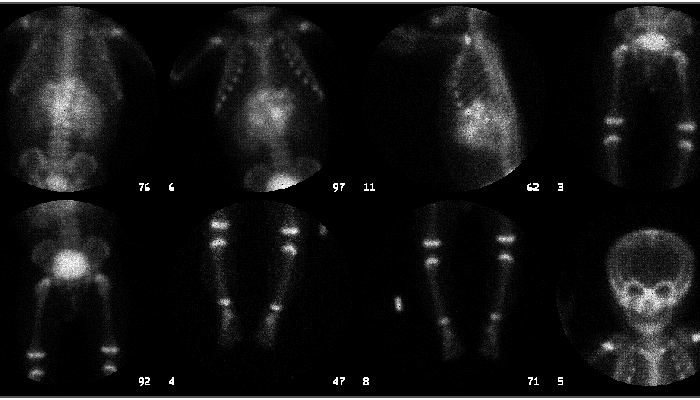
Bone scintigraphy
View main image(bs) in a separate image viewer
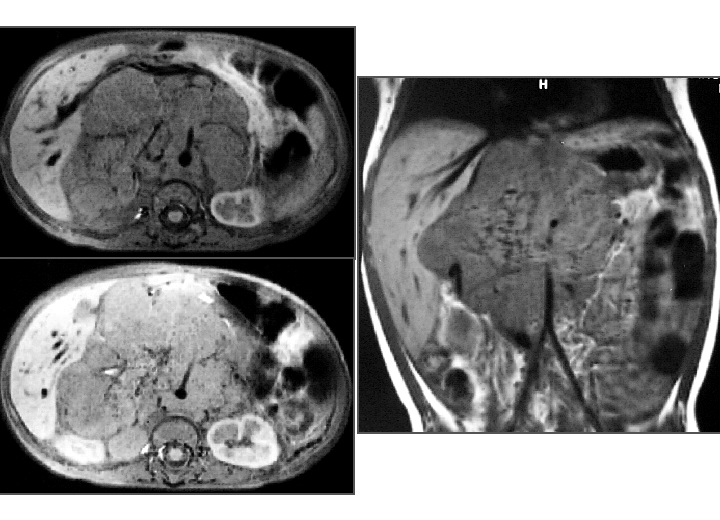
View second image(mr). MRI of abdomen
Full history/Diagnosis is available below
Extraosseous abdominal activity. No evidence of bone metastases.
2. MRI of abdomen (same day):
Large mid-abdominal mass, originating from the right adrenal gland.
The mass encases vessels (including the aorta, inferior vena cava, portal vein, celiac axis, SMA and renals).
Neuroblastoma accounts for 8 to 10% of all childhood tumors and has an incidence of approximately 8 per million per year in children under 15. Over 95% occur before age 10. The tumors have differentiation patterns similar to that of normal neuronal differentiation. Furthermore, neuroblastic nodules, or in situ neuroblastoma are found in autopsies of infants that died of other causes. It is unclear if these cell rests represent the origin of neuroblastic tumors, but it has been suggested that the tumors may result from error in cell differentiation. Neuroblastic tumors reveal the highest rate of spontaneous regression of all human tumors. They generally also have a very poor prognosis when presenting as disseminated disease. Two hypotheses exist regarding the pathologic differentiation of neuroblastic tumors. In one, the tumor is thought to progress from a relatively benign form to a more malignant form with time. In the other hypothesis, the tumor is thought to have a fixed character from the start and to not change with time. If the former is true, then screening programs designed to catch neuroblastic tumors while they are still in a relatively benign form should result in a reduced mortality rate. Several studies in such countries as Japan and Canada have not shown such a mortality benefit, lending credence to the latter hypothesis.
The international staging system for neuroblastoma is as follows:
Stage 1: Confined to area of origin. Complete gross resection, with or without microscopic residual disease. Lymph nodes negative.
Stage 2A: Unilateral with incomplete gross resection. Lymph nodes negative.
Stage 2B: Unilateral with complete or incomplete gross resection. Ipsilateral lymph nodes positive (contralateral negative).
Stage 3: Crosses midline with or without lymph nodes; or unilateral tumor with contralateral lymph nodes.
Stage 4: Distant metastases to lymph nodes, bone marrow, liver or other organs (except as defined in 4S).
Stage 4S: Localised primary (as defined in stage 1 or 2) with dissemination limited to liver, skin or bone marrow.
Three-year event free survival for patients of all ages with stage 1, 2 and 4S disease is 75 - 90%. For infants less than 1-year-old, it is 90% for stage 3 and 60 - 75% for stage 4. For children, it is 50% for stage 3 and 15% for stage 4. A variety of serum tests (e.g., ferritin, neuron-specific enolase, lactate dehydrogenase) have been hypothesized to have prognostic value, but are non-specific. Tumor genetic factors, such as N-myc copy number (N-myc is a proto-oncogene which is amplified in approximately 25% of neuroblastomas and is associated with poor prognosis), ploidy (tumor hyperdiploidy in infants is associated with a good prognosis), and deletions (especially 1p chromosome deletion, associated with a poor prognosis) provide important prognostic information.
Most patients with neuroblastoma undergo surgical resection and chemotherapy. Some patients also receive radiation therapy. Aggressiveness of therapy is dictated by the prognostic factors. Treatment with large doses of I-131-labelled metaiodobenzylguanidine (mIBG) is under investigation.
References:
1. Acharya, Suchitra et. al. Prenatally Diagnosed Neuroblastoma. American Cancer Society. 80: 2; 304-310, 1997.
2. Castleberry, Robert P. Biology and Treatment of Neuroblastoma. Pediatric Clinics of North America. 44: 4; 919-937, 1997.
3. Castleberry, R.P. Neuroblastoma. European J of Cancer. 33: 9; 1430-1438, 1997.
4. Favrot, M.C. Comparison of the Diagnostic and Prognostic Value of Biological Markers in Neuroblastoma. 7: 607-611, 1996.
5. Flower, Maggie A. and Fielding, Sue L. Radiation Dosimetry for I-131 mIBG Therapy of Neuroblastoma. Phys Med Biol. 41: 1933-1940, 1996.
6. Gaze, M.N. and Wheldon, T.E. Radiolabelled mIBG in the Treatment of Neuroblastoma. 32A: 1; 93-96, 1996.
Follow-up computed tomography three months later did not reveal a significant change in the size of the tumor. Calcification is apparent within the tumor.
References and General Discussion of Bone Scintigraphy (Anatomic field:Gasterointestinal System, Category:Neoplasm, Neoplastic-like condition)
Return to the Teaching File home page.
{kind=link}